Research at Kaelin Lab
Over time, cells can accumulate changes (mutations) in their DNA. If the DNA is a blueprint for making a protein (that is, a gene), the behavior of the cell might be altered. If the right combination of genes becomes altered, the cell will become cancerous. Some genes (tumor-suppressor genes) prevent malignant behavior. These genes contribute to cancer when they are inactivated as a result of mutations. Other genes (proto-oncogenes) can promote cancer if they acquire new properties as a result of mutations (at which point they are called oncogenes). Most common cancers involve both inactivation of specific tumor-suppressor genes and activation of certain proto-oncogenes.
Our laboratory studies the functions of the proteins encoded by specific tumor-suppressor genes. Our long-term goal is to help develop new anticancer therapies. For example, it might be possible to develop a drug that would mimic the behavior of a particular tumor-suppressor protein (for example, by inactivating a specific enzyme involved in cell growth). It might also be possible to design strategies for killing only those cells in which a particular tumor-suppressor protein has been inactivated (thus sparing normal cells).
The von Hippel–Lindau Tumor Protein
The hereditary cancer syndrome von Hippel–Lindau (VHL) disease is characterized by the development of multiple blood vessel tumors (hemangioblastomas) of the brain and eye, as well as tumors of the kidneys and adrenal glands. Individuals with VHL disease have inherited a mutated, or altered, VHL tumor-suppressor gene from either parent. Tumors develop when the remaining normal copy of the VHL gene (from the other parent) is inactivated, rendering the cell incapable of making the normal VHL protein (pVHL). VHL mutations are also very common in nonhereditary kidney cancers.
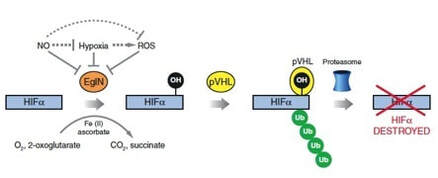
We and others previously showed that pVHL targets certain proteins for destruction by chemically linking them to a "flag" called polyubiquitin. This flag is recognized by a cellular machine, called the proteasome, that degrades proteins. The best-understood pVHL target is the protein HIF (hypoxia-inducible factor), implicated in the control of genes that are turned on by low oxygen. pVHL only targets HIF for destruction in the presence of oxygen. In the absence of oxygen (or in tumor cells lacking normal pVHL), HIF accumulates and activates genes that promote survival in a low-oxygen environment (such as occurs when the blood supply to an organ is compromised). Among these genes are genes that direct the synthesis of proteins that induce blood vessel formation, such as VEGF (vascular endothelial growth factor). We used laboratory models to establish that HIF is a driving force in the development of kidney cancer. Based on positive clinical trials, drugs that indirectly down-regulate HIF, such as inhibitors of the mTOR kinase, as well as drugs that inhibit VEGF (or its receptor KDR), have now been approved by the Food and Drug Administration for the treatment of kidney cancer.
We and others discovered that pVHL binds to a short, discrete region of HIF. At the heart of this region is a proline residue (1 of the 20 amino acid building blocks used to make proteins) that, in the presence of oxygen, becomes chemically modified by the addition of a hydroxyl group, leading to its recognition by pVHL. We purified the enzyme EglN1 (also called PHD2), which hydroxylates HIF in the presence of oxygen.
Laboratory experiments by us and others suggest that drugs that inhibit EglN1 would be useful for the treatment of anemia, heart attacks, and strokes. To facilitate the development of such drugs, we made a genetically engineered mouse in which tissues lacking adequate oxygen emit light, which can then be monitored using a sensitive camera, as well as mice in which EglN1 can be inactivated at different time points after birth.
Some VHL families develop adrenal gland tumors (pheochromocytomas) but do not develop hemangioblastomas or kidney cancers. (Pheochromocytomas are derived from the same cells that give rise to the sympathetic nervous system responsible for the fight-or-flight response). These families have VHL mutations that do not compromise pVHL's ability to recognize HIF. Instead, we discovered that these mutations affect a second function of pVHL that is important for killing excess cells during the embryological development of the sympathetic nervous system. We also showed that other genes linked to hereditary pheochromocytoma affect this same process. Escape from killing apparently sets the stage for these cells to form tumors later in life. These pheochromocytoma-related genes ultimately impinge upon EglN3, which is a "cousin" of EglN1. We found that killing by EglN3 requires the protein KIF1Bβ, which is encoded by a gene on the short arm of chromosome 1. This chromosomal region is frequently deleted in cancers such as pheochromocytomas and the pediatric sympathetic nervous system tumor called neuroblastoma.
Although VHL inactivation contributes to certain forms of cancer, it may also create therapeutically exploitable vulnerabilities. We inactivated, one at a time, all of the human genes that encode kinases (enzymes that add phosphate groups to other molecules) in matched cancer cells that do or do not have a normal version of the VHL gene. We have identified kinases that are more important for the survival of cells lacking VHL than in cells where VHL is intact. In theory, drugs directed against such kinases would preferentially kill cancer cells lacking VHL compared to normal, VHL-proficient, cells.
The Retinoblastoma Protein
The retinoblastoma tumor-suppressor protein (pRB) binds to a family of proteins collectively referred to as E2F. When not bound to pRB, E2F can turn on specific genes that stimulate cell growth. In contrast, these same genes are turned off once pRB binds to E2F. Hence, E2F means "go" and pRB means "stop."
Tumor cells in general, and cells lacking pRB in particular, frequently display defects in differentiation, the process whereby immature cells commit to perform specific functions. Our recent work suggests that control of differentiation by pRB is linked to its ability to inhibit the protein RBP2 (RB-binding protein-2). We found that eliminating RBP2 in cancer cells causes them to stop proliferating and to once again perform functions attributed to normally differentiated cells. To better understand RBP2's normal functions, we have now made mice that lack RBP2. Our preliminary data indicate that these mice are partially protected from cancers caused by pRB inactivation. DNA is normally associated with proteins called histones, which modulate how easy it is for the cell to "read" the information contained within the DNA and use it to make proteins. The functions of histones, in turn, are regulated by the addition or removal of small chemical entities such as methyl groups (a methyl group consists of one carbon atom and three hydrogen atoms). We recently discovered that RBP2 catalyzes (accelerates) the removal of methyl groups from histones under certain circumstances. This suggests that RBP2 regulates differentiation by modulating histone function.
Our laboratory studies the functions of the proteins encoded by specific tumor-suppressor genes. Our long-term goal is to help develop new anticancer therapies. For example, it might be possible to develop a drug that would mimic the behavior of a particular tumor-suppressor protein (for example, by inactivating a specific enzyme involved in cell growth). It might also be possible to design strategies for killing only those cells in which a particular tumor-suppressor protein has been inactivated (thus sparing normal cells).
The von Hippel–Lindau Tumor Protein
The hereditary cancer syndrome von Hippel–Lindau (VHL) disease is characterized by the development of multiple blood vessel tumors (hemangioblastomas) of the brain and eye, as well as tumors of the kidneys and adrenal glands. Individuals with VHL disease have inherited a mutated, or altered, VHL tumor-suppressor gene from either parent. Tumors develop when the remaining normal copy of the VHL gene (from the other parent) is inactivated, rendering the cell incapable of making the normal VHL protein (pVHL). VHL mutations are also very common in nonhereditary kidney cancers.
We and others previously showed that pVHL targets certain proteins for destruction by chemically linking them to a "flag" called polyubiquitin. This flag is recognized by a cellular machine, called the proteasome, that degrades proteins. The best-understood pVHL target is the protein HIF (hypoxia-inducible factor), implicated in the control of genes that are turned on by low oxygen. pVHL only targets HIF for destruction in the presence of oxygen. In the absence of oxygen (or in tumor cells lacking normal pVHL), HIF accumulates and activates genes that promote survival in a low-oxygen environment (such as occurs when the blood supply to an organ is compromised). Among these genes are genes that direct the synthesis of proteins that induce blood vessel formation, such as VEGF (vascular endothelial growth factor). We used laboratory models to establish that HIF is a driving force in the development of kidney cancer. Based on positive clinical trials, drugs that indirectly down-regulate HIF, such as inhibitors of the mTOR kinase, as well as drugs that inhibit VEGF (or its receptor KDR), have now been approved by the Food and Drug Administration for the treatment of kidney cancer.
We and others discovered that pVHL binds to a short, discrete region of HIF. At the heart of this region is a proline residue (1 of the 20 amino acid building blocks used to make proteins) that, in the presence of oxygen, becomes chemically modified by the addition of a hydroxyl group, leading to its recognition by pVHL. We purified the enzyme EglN1 (also called PHD2), which hydroxylates HIF in the presence of oxygen.
Laboratory experiments by us and others suggest that drugs that inhibit EglN1 would be useful for the treatment of anemia, heart attacks, and strokes. To facilitate the development of such drugs, we made a genetically engineered mouse in which tissues lacking adequate oxygen emit light, which can then be monitored using a sensitive camera, as well as mice in which EglN1 can be inactivated at different time points after birth.
Some VHL families develop adrenal gland tumors (pheochromocytomas) but do not develop hemangioblastomas or kidney cancers. (Pheochromocytomas are derived from the same cells that give rise to the sympathetic nervous system responsible for the fight-or-flight response). These families have VHL mutations that do not compromise pVHL's ability to recognize HIF. Instead, we discovered that these mutations affect a second function of pVHL that is important for killing excess cells during the embryological development of the sympathetic nervous system. We also showed that other genes linked to hereditary pheochromocytoma affect this same process. Escape from killing apparently sets the stage for these cells to form tumors later in life. These pheochromocytoma-related genes ultimately impinge upon EglN3, which is a "cousin" of EglN1. We found that killing by EglN3 requires the protein KIF1Bβ, which is encoded by a gene on the short arm of chromosome 1. This chromosomal region is frequently deleted in cancers such as pheochromocytomas and the pediatric sympathetic nervous system tumor called neuroblastoma.
Although VHL inactivation contributes to certain forms of cancer, it may also create therapeutically exploitable vulnerabilities. We inactivated, one at a time, all of the human genes that encode kinases (enzymes that add phosphate groups to other molecules) in matched cancer cells that do or do not have a normal version of the VHL gene. We have identified kinases that are more important for the survival of cells lacking VHL than in cells where VHL is intact. In theory, drugs directed against such kinases would preferentially kill cancer cells lacking VHL compared to normal, VHL-proficient, cells.
The Retinoblastoma Protein
The retinoblastoma tumor-suppressor protein (pRB) binds to a family of proteins collectively referred to as E2F. When not bound to pRB, E2F can turn on specific genes that stimulate cell growth. In contrast, these same genes are turned off once pRB binds to E2F. Hence, E2F means "go" and pRB means "stop."
Tumor cells in general, and cells lacking pRB in particular, frequently display defects in differentiation, the process whereby immature cells commit to perform specific functions. Our recent work suggests that control of differentiation by pRB is linked to its ability to inhibit the protein RBP2 (RB-binding protein-2). We found that eliminating RBP2 in cancer cells causes them to stop proliferating and to once again perform functions attributed to normally differentiated cells. To better understand RBP2's normal functions, we have now made mice that lack RBP2. Our preliminary data indicate that these mice are partially protected from cancers caused by pRB inactivation. DNA is normally associated with proteins called histones, which modulate how easy it is for the cell to "read" the information contained within the DNA and use it to make proteins. The functions of histones, in turn, are regulated by the addition or removal of small chemical entities such as methyl groups (a methyl group consists of one carbon atom and three hydrogen atoms). We recently discovered that RBP2 catalyzes (accelerates) the removal of methyl groups from histones under certain circumstances. This suggests that RBP2 regulates differentiation by modulating histone function.